Clovis Oncology (CLVS)
Execution Risk, Loss of Credibility, & Regulatory Missteps Constrain Valuation. Better Opportunities Elsewhere.
November 27, 2015
Background:
We held a call with Clovis Oncology on November 18 in the days after its announcement revealing that Rociletinib's true efficacy was about half of what was previously reported to investors the week prior despite having this data in-house since October. It also stated that the FDA requested "additional data," during its mid-cycle NDA meeting held in the middle of October and that it would file a "major amendment" which management stated that this consisted of "line-by-line patient data." However, after our call with CLVS we came away feeling less confident with more questions remaining than they were willing to answer, and believe valuation will suffer under this crisis of confidence into perpetuity as long as current management remains in place. It is extremely difficult to come back from such a blow to confidence especially when management marketed itself prior to its IPO as a team that will "focus on clinical development and managing the regulatory interface so critical and often delayed too long." CLVS' clinical & regulatory development strategy has proven to be impetuous and the probability of approval is not equivalent to probability of reaching peak sales. This is a mistake we frequently see from both the buy-side and sell-side.
Two Key Tidbits from the call were:
(1) CLVS expects to hear from the FDA by November 30, on whether or not the FDA will accept its "major amendment" to Rociletinib's NDA package that was filed on the evening of Monday, November 16. This represents a potential downside risk in the near term if it receives a complete response letter (CRL) from the FDA;
(2) Rociletinib's commercial success hinges on future drug combinations. Based on Management's reformed view on Roci's commercial potential, it appears to us (and management confirmed this) that the only realistic way to catch up on efficacy to AZN's Tagrisso is through new drug combinations.
The thesis has since transformed into one where the approval of Rociletinib is now questionable, and even if approved unlikely to gain significant market share commercially (<40% market share) and we would argue that Rociletinib's pricing power is weak on par with Tarceva's list price and would have to be priced at a 30-50% discount to Tagrisso's list price of $12,750. Prior to these developments, the thesis was that Rociletinib would launch during the 4Q15 or 1Q16 and would secure 50-70% market share in the T790m+ setting, but this was based on its supposed superior clinical profile relative to AZN's Tagrisso. The thesis was also supported by the fact that CLVS could have its second product approval with Rucaparib for gBRCA+ ovarian cancer in 2017. The probability of such an outcome occurring now is <5%.
- Recall, we were very bullish on CLVS since November 19, 2014 (ref. entry price $48.28), and saw peak gains of +142% at the all-time high of $116.65 on September 18, 2015. Since then shares are down more than -70%. We have considered a multitude of potential outcomes for Rociletinib and Rucaparib and believe that fair value is between $21-$30 per share, while our bull case now is $43. Note in November 2014, our bear case was $38 per share.
- When the facts change everything changes: What Rociletinib represented in October, is not what it is today, good contrarian trade setups must be based on companies with solid fundamentals; CLVS no longer possesses these attributes.
- On November 25, Hayman Capital Management, LP published a semi-bullish note on CLVS with a base case price target of $45 primarily based on a contrarian view and hope for a dead cat bounce. In our view, this is biotech tourism at its best, identifying a potential contrarian trade idea hoping for a "dead cat bounce" while completely neglecting to account for the new clinical, regulatory, and commercial realities for Rociletinib. We get their thesis. Sentiment is poor, and stock just blew up, in theory a good risk-reward.
- We see the reward-risk from current levels (~$30 per share) of +50% to the upside & -50% to -70% to the downside, and despite shares trading down -70% this month, the risk-reward remains unfavorable here at ~1:1. We prefer to wait until the other shoe drops on Rociletinib's PDUFA date or complete response letter from the FDA where we could see shares trade down toward $15-$20.
- We would sidestep Mr. Bass's recommendation to enter long CLVS here and wait for a move lower to $15-$20, or until CLVS hears back from the FDA on its NDA status. Should CLVS receive a CRL, we see shares rapidly trading down toward $15 per share. With both the quality of management and the quality of data (and data drives valuation not sentiment) justifiably being called into question the risk-reward on CLVS does not represent a compelling asymmetric risk-reward in light of the new realities.
- CLVS remains in a precarious position fundamentally, and requires a thorough understanding of how the true clinical data has changed everything. We estimate CLVS has two to three years of operating expenses in cash and with an unknown amount of legal liabilities, cash burn becomes increasingly important.
Caution on Hayman Capital's Bullish Model on CLVS
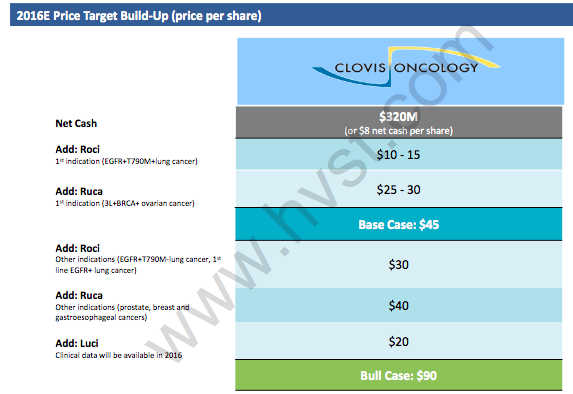
On November 25, the notable portfolio manager Kyle Bass of Hayman Capital Management, LP published a semi-bullish note on CLVS with a price target of $45 primarily voicing their contrarian view and hope for a dead cat bounce.
- Their base case price target is about on par with our revised model bull case scenario of $43 per share, but is widely divergent from our base case of $30 fair value for Rociletinib and Rucaparib. While their base case valuation is rational, it fails to account for management's loss of credibility, heightened regulatory & commercial risk. We believe that near to intermediate term data updates are highly unlikely to catalyze shares as they have done in the past with many trapped longs (new shorts) likely selling any meaningful "pop" in the stock. Moreover, if CLVS doesn't stem its cash burn, the "cash value" of the company will erode, and insolvency is possible should Rociletinib not get approved catalyzing "strategic alternatives" (read: layoffs, asset sales, and out-licensing) to sustain operations.
Hayman Capital's bull case of $90 has <15% probability of actually occurring in our view, with 44% ($40) coming from Rucaparib that include indications where no clinical data exists, and 22% ($20) from Lucitanib. We note CLVS already discontinued Rucaparib in pancreatic cancer, and it's unwise to even contemplate attributing any valuation from indications outside of gynecological malignancies especially given the perception issues at CLVS. As we stated previously, keen biotech investors are unlikely to assign value as they have in the past to new data releases going forward. Definitively, we know that gaining clinical validation in these new indications only intensifies its cash burn and given the current uncertainty those programs are unlikely to have validation for several years. We remain cautious of any thesis that changes so drastically to the downside that now appears to be fully dependent on aggressive modeling assumptions to fill the void in valuation left from Rociletinib's dissipating commercial opportunity.
In our November 2014 report, we were the first ones to highlight the baseline differences in race between CLVS and AZN trials while the Street only awoke to this at ASCO 2015. However, the fact remains that the FDA did not care enough about this to include any language on the Tagrisso's labeling thus downgrading its relevance as a moot point. Moreover, in all clinical trials there were only numerical trends suggesting that Asians had a more favorable response to EGFR TKI's such as Tarceva or Iressa, none of the data were statistically significant and hence moot, not clinically or commercially meaningful. Moreover, most oncologists do not know or believe the magnitude of effect is significant enough to risk using an inferior drug on efficacy on the speculative notion that there is a statistically significant and real clinical differences based on baseline patient differences on race. This cannot be assumed unless head-to-head clinical trials are done with Roci vs. Tagrisso. The magnitude of difference on ORRs between Roci and Tagrisso is so profound that racial disparities are unlikely to be the sole culprit, to suggest otherwise at this point in the game only confirms our skepticism of management's credibility. To attempt to minimize this steep drop in ORRs as "botched optics" in study design is undeniably spinning bad data into decent data. If the disparity between confirmed vs. unconfirmed was only 5-15% racial disparities could be responsible, but the delta is so wide, only one conclusion can be made… Rociletinib is inferior. If CLVS believes that race is the cause of the disparity. PROVE IT with head-to-head studies. The burden of proof on all matters is now on them.
We humbly ask:
- How did Hayman Capital model clinical development expenses for these new indications for which they assign so much value for Rucaparib?
- Was it in addition or in-lieu of Roci's and Rucaparib's existing clinical development commitments & expenses, which according to our call with CLVS all trials remain on track.
- How will CLVS fund these studies if Rociletinib approval is delayed/denied?
- Higher operating expenses will be required to validate these indications clinically, and with Roci's future looking grim, timelines are likely to be very slow. Recall, higher Opex decrease the NPV of FCF and without a thorough description from Hayman Capital on their model assumptions, their "base case" and "bull case" should be viewed with caution.
We channel Howard Marks and believe investors need to exercise "second-level thinking" here with CLVS and must consider the probabilities of second, and third order outcomes. Hayman Capital argues, "Rociletinib is still approvable" and "Roci usage in its initial indication may be higher than the market anticipates because doctors recognize the difference in study populations used in Roci's (Clovis) clinical trials vs. Tagrisso's (AstraZeneca) clinical trials" (no they really do not care that much given the -25-30% disparity in confirmed ORRs). We have spoken with multiple oncologists stating, "Roci is commercially dead" and "oncologists aren't going to analyze waterfall plots and use subgroup analyses when confirmed ORRs are half of Tagrisso's."
Hayman Capital's bullishness also hinges on old data from Rociletinib's efficacy in the T790m- population of 37% ORR (unconfirmed) will not dissipate another 30%-50% lower as the confirmed ORRs did in T790m+ data has. In light of recent events, trusting any data beyond confirmed ORRs is risky; these numbers are likely to fall too. In our most humble opinion, this represents first level thinking, when the quality of data is called into question everything changes, and until CLVS has complete transparency and only provides confirmed response rates going forward, we cannot trust any of its past data or assess its competitive positioning.

Hayman Capital Model (source: HVST)
The data is the data, and commercial planning & marketing strategies will not change the commercial disadvantages if approved. When the facts change, the thesis changes, and when management perpetuated misleading data despite knowing otherwise, we cannot give them the benefit of the doubt a moment longer.
The dramatic reduction in Rociletinib's commercial opportunity exclaims the hidden truth here…
- It makes very little sense on an NPV basis to carry the heavy cost of commercialization alone and believe partnering Rociletinib ex-US makes a lot of sense to generate some cash, and reduce future cash burn.
- Should our bear case on Rociletinib play out that assumes only 40% market share in the T790m+ 2nd and 3rd line setting, and 0% in the front-line EGFR+ setting, Rociletinib carries a -$6 per share in NPV for CLVS.
- Should CLVS fail to take action, it has ~2 years of cash to fund operations assuming ~$300M per year. If Rociletinib's PDUFA is delayed or CLVS receives a complete response letter from the FDA the risk of insolvency even upon commercial approval of Rucaparib in 2017 would be unlikely to provide a large enough sales base to offset this risk.
- Barring Rociletinib gaining approval as originally hoped by March 30, 2016, we see CLVS needing to raise additional equity or debt, if it even could at this juncture with current management.
Drug development is inherently risky, we all are aware of this.
However, when management has possibly maintained a facade and confidence in management has been shaken to this degree, lingering doubts are likely to remain on their openness with investors for quite some time, and its valuation is unlikely to recover meaningfully. Furthermore, how will the outcome from shareholder lawsuits impact their financial positioning, and perhaps the more beckoning question, how will this impede their access to capital over the next two years?
Consequently, the stock is likely to remain a very contentious bear story and any upside to valuation from future pipeline success is unlikely to be given much value by investors. Now with the potential for M&A vanished, bears remain strongly in control with just cause.
Our impression and valuation of CLVS has changed, and we believe a cloud of doubt over management's credibility will remain indefinitely. Their actions over the past two months are very questionable, and caught the market (and us) completely off-guard. Management has always touted itself to be experienced with "clinical development and managing the regulatory interface," but the recent events suggest otherwise. In a rush to catch-up to AstraZeneca (AZN) management made significant missteps with their NDA filing for Rociletinib that are only now coming to light, and breached its trust with investors. Recall, we were very bullish on CLVS since November 19, 2014 (ref. price $48.28), and saw peak gains of +142% at the all-time high of $116.65 on September 18, 2015. Since then shares are down more than -70%.
We see reward-risk from current levels (~$30 per share) of +50% to the upside & -50% to -70% to the downside, and despite shares trading down -70% this month, the risk-reward remains unfavorable here at ~1:1. We prefer to wait until the other shoe drops on Rociletinib's PDUFA date or complete response letter from the FDA where we could see shares trade down toward $15-$20.
The consequences of their regulatory missteps may not be over, and there could be additional pressure on shares as soon as next week. During our call with CLVS, it stated that it expects to hear back from the FDA by November 30th on whether or not the FDA will either:
- Extend Rociletinib's PDUFA date a minimum of the 90-day average (~Late June/early July)
- Send CLVS a complete response letter on the basis of an incomplete NDA submission (by November 30th according to management, delays Roci approval until at least 2017, if ever).
- Maintain the current PDUFA March 30th, 2016 (least likely, but commercial potential of Roci has dramatically deteriorated) with recommendations on labeling that excludes patient with baseline CNS metastases
CLVS continued to present old data to investors on Rociletinib's unconfirmed objective response rates (ORR) in lung cancer (NSCLC) despite having the new (confirmed ORRs) data in-house since the end of October. The delta (change) between what was communicated to the street this month (50-60% ORRs), and what is now being told to the street (28-34%) was a difference of 20-30% between unconfirmed (1 scan) and confirmed responses (1 scan + 1 follow up scan) showing at least -30% tumor regression by RECIST criteria. The response rates were clearly different and this dramatically changes the market opportunity for Rociletinib.
The bottom line:
The new clinical profile for Roci is ½ the response rate compared to AZN's Tagrisso, with lower incidence of rash and pneumonitis/ILDs, but with high risk of hyperglycemia, which we used to not view as a major negative, but that was only in the context of equal or better than ORRs relative to Tagrisso. This is no longer the case. When the facts change, the thesis changes, and when management perpetuated misleading data despite knowing otherwise, we cannot give them the benefit of the doubt a moment longer.
Conceivably, Rociletinib could fill a small niche of the market in certain populations such as patients at risk of severe rash, or increased risk of interstitial like lung events (ILDs) , but "oncologists will not analyze spider plots of data, or splice the data between unconfirmed versus confirmed responses, they will go with what's on the FDA label, and Tagrisso has the better label" according to our oncologist who warned that "oncologists are not going to be persuaded by some sales rep."
Others echoed these views as well. According to the head of Translational Medicine "AZN and CLVS was the classic horse race in drug development, AZN had the better molecule, once daily dosing vs. Rociletinib's twice-daily dosing, and now we know AZN's drug had better ORRs. Rociletinib lost time to market; perception has now soured, and is "commercially dead."
We also spoke with several consultants with experience in FDA regulatory submissions. For commercial timelines, the minimum consequence of the recent data disclosure is that Rociletinib's PDUFA date will likely be extended 90-days from their original PDUFA date scheduled for March 30, 2016. This would imply an optimal scenario a new PDUFA date from the FDA toward the end of June or mid-July. Furthermore, CLVS stated that it "should" hear back from the FDA on an updated PDUFA date before November 30, which could also act as the proverbial "other shoe to drop" on CLVS' valuation already dwindled down to an enterprise value of <$1B from more than $3B in October.
CLVS knew the data was materially different than its previous communiqués with the market and had the revised data in-house since mid-late October, but chose to delay informing its investors during two subsequent investor updates including its 3Q15 earnings call on Nov. 5 & Credit Suisse's Global Healthcare Conference on Nov.11. During its ASCO 2015 investor presentation in June, it presented data on unconfirmed response rates without noting this anywhere on the presentation itself. Similarly, in September, it reported the same ORRs at the World Conference on Lung Cancer Conference (WCLC).

In their defense, management frequently stated that their data is still "immature," but this usually is in reference to endpoints such as progression-free survival (PFS). Their logic in delaying this negative investor update does carry some plausible deniability, stating only that they were waiting to hear from the FDA before informing their investors. Our current view is that CLVS "wanted to definitively know if the FDA would consider both confirmed and unconfirmed for Rociletinib's approval before making an announcement."
However, the regulatory standard is to consider only confirmed response rates and this was an unambiguous fact from the FDA. So, how can an "experienced" management team expect special treatment or exemptions from regulatory standards? Any rational investor knows that this fails the simple "smell test." They knew, and delayed informing their investors.
To review briefly, patient data was submitted in a pooled analysis from TIGER-X and TIGER-2, now showing that confirmed objective response rates of only 28% at 500mg (n=79) and 34% at 625mg (n=170) compared to AZN's 59% ORR on Tagrisso's (AZD-9291) newly approved FDA label. Recall, CLVS was reporting unconfirmed response rates by RECIST criteria between 54-60% as recent as November 11 to investors despite knowing the confirmed response rate was much lower mid-late October. Consequently, if approved, sales are unlikely to begin until 4Q16 or 2017 in a best-case scenario
In retrospect was the bad data obvious?
- On September 3, 2014, Clovis announces a $250 million aggregate principal amount of its 2.50% convertible senior notes due 2021.
- NDA submission on July 1, 2015.
- Secondary equity offering on July 7th, pricing 4,054,4871 shares of its common stock at $78.00 per share, raising approximately $318M, inclusive of underwriter 30-day option.
- On June 22nd, 2015, CMO leaves during the summer unexpectedly,
- Followed by the resignation of its CFO on November 5, 2015.
Perhaps we are speaking for the market too much?
But, these are the developments we would need to see to ever regain confidence in the company, the drug, and its market potential. Because the burden of proof remains heavily on CLVS now, the only data that will alleviate this burden of doubt from the market's perspective will be survival data (PFS and/or OS), which requires prolonged timelines.
What this means for the stock?
Time decay to valuation, and any strong rallies are likely to be sold down by trapped longs and short-sellers re-entering. Additionally, new ORR data from Rociletinib will unlikely act as a meaningful catalyst to shares for the foreseeable future, and investors will need to see mature PFS and overall survival (OS) data before regaining confidence in Rociletinib's commercial potential.
The Principles of Pharmacology in Cancer Combination Therapy May Provide the One Ray of Hope for Rociletinib Going Forward…
The essential principle of pharmacology in cancer combination therapy is to combine two or more agents with complementary mechanisms of action, but have minimal overlapping toxicities. The one potential saving grace for Rociletinib commercial potential is if confirmed ORRs with combination therapies prove to be equivocal or better than monotherapy with Tagrisso, with minimal additional toxicity, and with superior overall survival.
With Rociletinib's seemingly inferior efficacy profile relative to AZN's Tagrisso, the last remaining differentiator for Rociletinib remains its safety profile with no patient deaths (yet) reported from interstitial like lung events (ILDs).
Our previous bullish thesis from 2014 was established on the following themes:
- Rociletinib had a superior safety profile and potentially a better efficacy profile compared to Tagrisso, that at worse would be equivalent to marginally inferior as data matured, and thus would secure more market share.
- Roci's superior safety profile would best be suited for future drug combinations with PD/L1 inhibitors and other agents.
- High probability of M&A: If Rociletinib was efficacious enough to secure frontline EFGR+ approval it would then represent a lifecycle management solution for Roche's Tarceva that loses patent exclusivity in 2018.
We argued in 2014, that Rociletinib's better safety profile was uniquely suited in combination with checkpoint inhibitors, especially in NSCLC because one of the prominent toxicities with immuno-oncology checkpoint inhibitors is inflammation and in NSCLC pneumonitis (lung inflammation) would be an on target toxicity. Obviously, this is a horrible toxicity in lung cancer patients, which is why we viewed Roci's safety profile as superior to Tagrisso, because we saw the long term potential for Roci in combination with MRK's PD1 inhibitor pembrolizumab, and Roche's anti-PDL1 inhibitor atezolizumab that will begin enrollment 4Q15/1Q16.
As a result, when CLVS began announcing collaborations with Roche, and then MRK on combo trials, our confidence grew that the true value potential with Roci was with immuno-therapy, and this drove both our thesis and M&A valuation. Roci's toxicity profile primarily consists of hyperglycemia, which is generally a benign side effect for lung cancer patients who are unlikely to live more than 1-3 years. But the real value potential was due to its differentiation on less lung toxicity and carried a very low incidence of ILDs ~1.5% with no patient deaths compared to AZN's ~3.3% incidence of ILDs and 4 patient deaths. That is 2x the risk of lung events with Tagrisso. We gained confidence in this thesis when AZN halted two Clinical Trials, the phase III CAURAL and phase I TATTON trial that revealed an increased incidence of interstitial lung disease (ILD) in patients who were taking Tagrisso + Durvalumab (AZN's anti-PDL1 inhibitor). These developments increased our confidence further that Roci's safety profile was better than Tagrisso's in combination PD1/L1 inhibitors and thus we saw an increasing probability of M&A. AZN noted it has not seen ILDs with Durvalumab monotherapy. ).
Overall, the wide divergence between unconfirmed and confirmed objective responses by RECIST criteria suggests that durability of responses appears to be the fundamental weakness with Rociletinib. Consequently, the only potential upside scenario for Roci from here that will not play out for at least another 12-18 months, would be clinical success of Rociletinib with PD1/L1 inhibitors, where it appears that AZN has failed so far, with two clinical trials being halted with Tagrisso + Durvalumab. We suspected since 2014, that Tagrisso's tox profile violated one of the fundamental principles of pharmacology in oncology, "don't use two drugs with overlapping toxicity."
Our view was confirmed, with CLVS' management stating that "drug combinations with Rociletinib have obviously become a priority" indirectly intimating to us that they will likely need to have clinical success in combination with other therapies in order to remain competitive commercially. The first Rociletinib data in combination with checkpoint inhibitors could be presented at ASCO (2Q) or 3Q/4Q16 at a European Conference (ESMO).
But, will the market even attribute any future NPV to this early stage data, given the loss of confidence?
It's a safe assumption that it will not. It's important to note that if CLVS receives a CRL from the FDA that second NDA submissions have historically had higher probabilities of success. CLVS is expected to file its NDA for Rucaparib during the 1H16, but the market is unlikely to apply a high probability of success or market share now given CLVS' seemingly amateur missteps with Rociletinib.
Should CLVS receive a CRL or PDUFA date extension, which is expected by November 30, we think that the optimal course of action for CLVS is to implement the following initiatives:
- CLVS is positioned to waste a lot of shareholder capital on a sales force that may not be needed until 2017. And based on our discussions with them they have no plans on "right-sizing" their commercial team "until they hear a 'no' from the FDA." Given Rociletinib's late stage, and relatively clean safety profile its makes increasing sense for CLVS to explore out-licensing Rociletinib ex-US (maybe even globally) and stop hemorrhaging shareholder capital (~$568M raised since 2014) by laying-off most of its sales force to reflect the diminished market opportunity in the 2nd/3rd Line T790m+ monotherapy setting and supplement with a contract sales force IF Rociletinib is approved at all in 2016.
- Ideal partners would be either Roche or NVS, both of which will begin enrollment with planned combination trials in the 4Q15 or 1H16.
Changes to Our Model to Reflect the New Realities:
- We brought down probabilities of success, market share, and treatment durations to reflect the new data (worse data).
- We now model sales starting in 2017E in 2L/3L T790m+ setting only that are risk adjusted at 65% (from 95% upon NDA filing) and in the front line setting sales are risk adjusted at 50% (from 75%) starting in 2018E.
- We note that drug pricing was also reduced to only $6,500 per month growing 1% per annum from 3% previously. This compares to Tagrisso's gross pricing of $12,750 per month.
- Peak global sales in our base case scenario Rociletinib now stand at ~$500-$550M fully risk-adjusted and discounted at a 10% WACC.
- This produces a fully diluted NPV of $20-$21 per share including cash, debt, and no value from Rucaparib or Lucitanib which we apply 0% probability of success here in this analysis.
- Base Case ($550M peak sales): We model 30% market share in front line setting, and 40% in 2nd and 3rd line setting. I
- Bear Case (<$200M peak sales): If we assume NO APPROVAL in the front line setting and 40% market share in second and third-line setting. Rociletinib is worth $5 per share.
- Bull Case ($1B Peak Sales): Roci is approved in the front line and 2nd and 3rd line setting with 50% market share in both lines of treatment, but with our reduced probabilities of success, reduced treatment durations, and lower drug pricing the NPV is still only $31 per share. This is highly unlikely to happen.
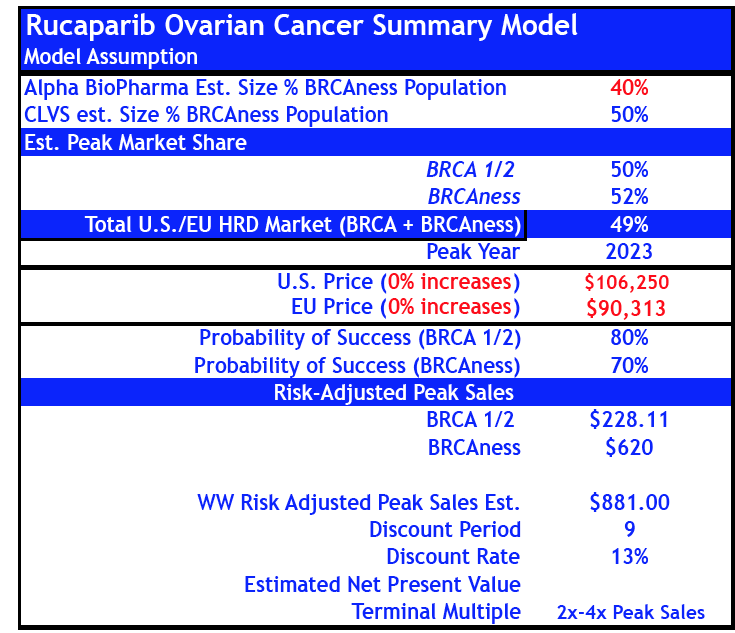
- For Rucaparib, we reduced probabilities of success by -50% in gBRCA+ and "HRD+ BRCAness" ovarian cancer from 80% and 70% to 40% and 35% respectively.
- If we include revised sales estimates for Rucaparib to our base case Rociletinib model peak sales increase to $833M by 2024E.
Revised Rucaparib Model (reduced probability of success)
Previous Rucaparib Model (May 07, 2015)
- As a separate analysis, we evaluated M&A takeover scenarios not because we think this is likely to occur anymore, but as a separate valuation model for investors who prefer to think of valuation using multiples of sales instead of DCF models. Overall, the true clinical profile of Rociletinib dramatically reduces M&A valuations and probabilities. In our Bull Case sales scenario with ~ $1.1B in peak global sales applying a 5x peak sales multiple has a $43NPV discounted annually at the 10% WACC. While combining our Bear Case for Rociletinib and Bear Case with Rucaparib only produces a takeover valuation of $17-$21 per share using a 4x-5x multiple to peak sales.
Know when the Rules to the Game have Changed…
Rociletinib was granted Breakthrough Therapy designation by the FDA in May 2014. On September 29, 2015, FDA Grants Priority Review Status to Rociletinib New Drug Application; Assigns Action Date (PDUFA) of March 30, 2016
To qualify for this designation, a drug must:
- Treat a serious or life threatening disease or condition
- Provide preliminary clinical evidence indicating a potential for substantial improvement over existing therapies on one or more clinically significant endpoints
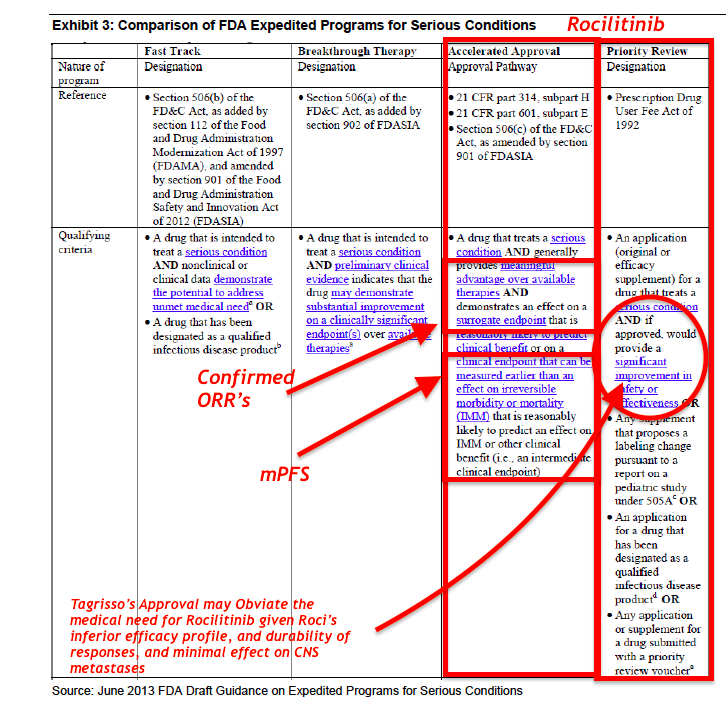
What Rules have Changed?
The Regulatory Hurdle for Approval has gone up…and Competitive Profile Dramatically Weakened…
- When the FDA granted Breakthrough Therapy designation (BTD) for Rociletinib, it was based on unconfirmed responses that were almost 2x the confirmed responses disclosed this month.
- Moreover, we question if the FDA might revoke Rociletinib's BTD status, as it clearly does not represent an "improvement over existing therapies." Yet, in Hayman Capital's bullish assessment, they completely ignore these new risks to Rociletinib's pathway to approval, and from the slide presentation it appears they swallowed management's "party line" whole without any rigorous academic debate.
- We prefer to wait for clarity from the FDA on whether Rociletinib's NDA is even approvable in its current form.
- Hence, as we stated at the beginning of this report, "what Rociletinib was then, is not what it is today…when the facts change, so does the thesis."
- We believe the recent reset of expectations on CLVS is justifiable and reflects important realities about management's legitimacy and fundamental truths about Rociletinib's regulatory and commercial outlook. It is unlikely that poor investor sentiment is transient in nature, but more likely a permanent stain on the company. At this point, with all publicly available information, the risk-reward is balanced, not asymmetric, and we prefer to wait until CLVS trades below $20 before considering going long. At $30 per share, CLVS already reflects Rociletinib approval with reduced commercial potential, and Rucaparib approval in gBRCA+ ovarian cancer.
Brief Overview on FDA "Major Amendments" to NDA Filings
The FDA can move the PDUFA date when there is a "major amendment" made to an NDA which can be because the FDA has requested certain information (a solicited amendment) or because the sponsor of the compound submits an amendment because there is new data that has to go into the application, such as results from a study (an unsolicited amendment). CLVS submitted a "major amendment" on Monday evening November 16th, 2015 that included "line-by-line patient data." Examples that constitute a "major amendment" are information about:
- Protocol deviations and the impact that they might have had on study outcomes;
- That provides clarification about a manufacturing site;
- Data inadvertently left out of the final study report;
- There is new validation data and analyses;
- Updated data from a previously reported study.
However, just because there is a major amendment doesn't dictate that the PDUFA date will have to be extended, it just means that it can. If the major amendment is submitted early enough (>90 day before PDUFA) in the review cycle then the division overseeing the application may not choose to extend the date. If however, the information is submitted to the FDA within 90 days of the PDUFA date, then the original PDUFA date will be extended. In either case, the FDA can make any decision they choose here, the data CLVS submitted to the FDA in October during its mid-cycle review meeting (90-days post NDA acceptance for review) showing significantly reduced confirmed ORRs may lead to a complete response letter on grounds of an incomplete NDA package, and possibly lead to the FDA revoking its BTD status.
Overview of FDA Policies and Procedures for "Major Amendments" to an NDA
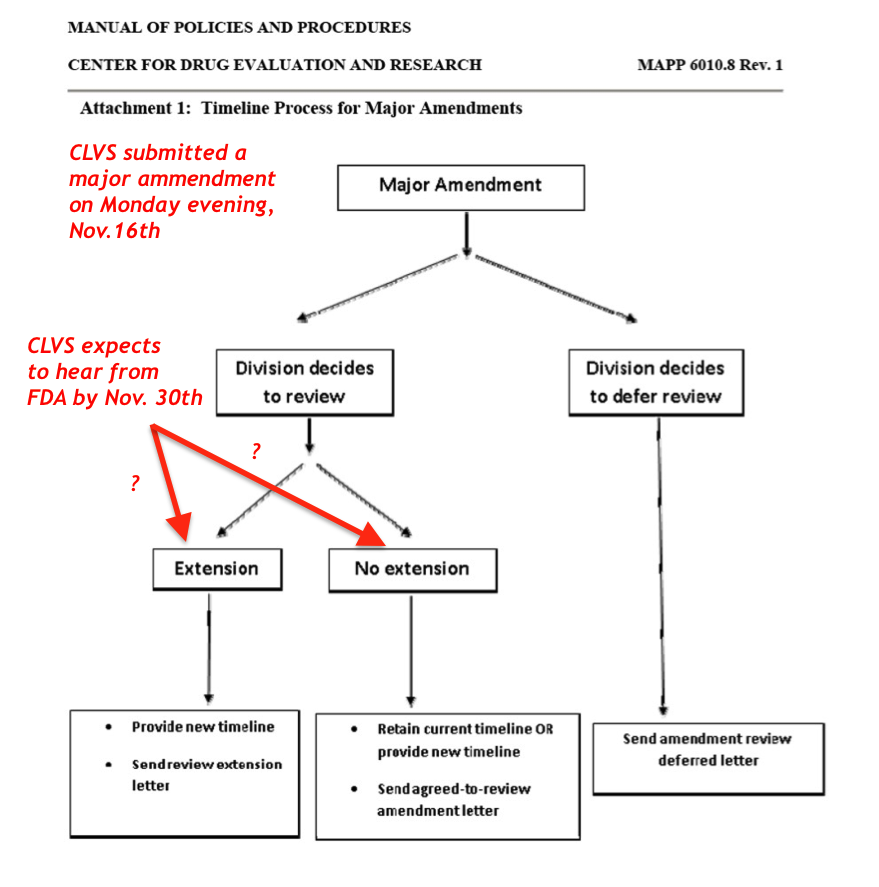
Complete Response Letter
We briefly provide some key insights into what a CRL is, what the subsequent options would be for CLVS if it receives one from the FDA as well as some important aspects that may pertain to CLVS' NDA for Rociletinib after its submission of a "major amendment" on the evening of Monday, November 16. Recall, CLVS expects to receive a response from the FDA by November 30; however, it was not made clear what this communication from the FDA will consist of. Conceivably, it could simply be a notification that the FDA is "accepting" its major amendment for review, notice on a new PDUFA date, or potentially a CRL.
The FDA will send the applicant a complete response letter if the agency determines that they will not approve the application or abbreviated application in its present form for one or more reasons.
In our view, the outcome of CLVS' 90-day mid-cycle review meeting with the agency requesting additional data is a clear indication that Rociletinib's NDA package provided "Inadequate data." Under this definition in the FDA's draft guidance on CRLs "if the FDA determines, after an application is filed or an abbreviated application is received, that the data submitted are inadequate to support approval, the agency might issue a complete response letter without first conducting required inspections and/or reviewing proposed product labeling."
There are a range of potential outcomes for Rociletinib's NDA review timelines, our base case assumption is that the FDA will delay its review by 90-days, but we would not be at all surprised if Rociletinib's received a CRL. This does not mean we think that Rociletinib is not approvable, but with the current efficacy data, the net clinical benefit has become highly questionable in our view given Roci's inability to control CNS metastases.
Should CLVS receive a CRL, the FDA will provide a "recommendation of actions for approval. When possible, a complete response letter will recommend actions that the applicant might take to place the application or abbreviated application in condition for approval." If there are any actions CLVS could take the most likely is simply waiting for data to mature on mPFS. At which point, CLVS would then be required to take one of the following actions:
- Resubmit the application or abbreviated application, addressing all deficiencies identified in the complete response letter.
- A resubmission of an application or efficacy supplement that FDA classifies as a Class 1 resubmission constitutes an agreement by the applicant to start a new 2-month review cycle beginning on the date FDA receives the resubmission.
- A resubmission of an application or efficacy supplement that FDA classifies as a Class 2 resubmission constitutes an agreement by the applicant to start a new 6-month review cycle beginning on the date FDA receives the resubmission.
- A resubmission of an NDA supplement other than an efficacy supplement constitutes an agreement by the applicant to start a new review cycle the same length as the initial review cycle for the supplement (excluding any extension due to a major amendment of the initial supplement), beginning on the date FDA receives the resubmission.
- (A major resubmission of an abbreviated application constitutes an agreement by the applicant to start a new 6-month review cycle beginning on the date FDA receives the resubmission.
- A minor resubmission of an abbreviated application constitutes an agreement by the applicant to start a new review cycle beginning on the date FDA receives the resubmission.
- Withdrawal. Withdraw the application or abbreviated application. A decision to withdraw an application or abbreviated application is without prejudice to a subsequent submission.
- Request opportunity for hearing. Ask the agency to provide the applicant an opportunity for a hearing on the question of whether there are grounds for denying approval of the application or abbreviated application under section 505(d) or (j)(4) of the act, respectively. The applicant must submit the request to the Associate Director for Policy, Center for Drug Evaluation and Research, Food and Drug Administration, 10903 New Hampshire Ave., Silver Spring, MD 20993. Within 60 days of the date of the request for an opportunity for a hearing, or within a different time period to which FDA and the applicant agree, the agency will either approve the application or abbreviated application under 314.105, or refuse to approve the application under 314.125 or abbreviated application under 314.127 and give the applicant written notice of an opportunity for a hearing.
We also reviewed data collected by the FDA and presented in a presentation: "Breakthrough Therapy Designation: Exploring the Qualifying Criteria" to assess if Rociletinib still meets eligibility for BTD status and approval. In our view, the new data from Rociletinib is substandard to existing therapies (Tagrisso). Specifically, we highlight the fact that the FDA assesses the difference between the primary endpoint value and the value from most effective available therapy to approximate therapeutic benefit. In this case, confirmed ORRs is the key variable to assess. As we know, CLVS is 25-31% inferior to Tagrisso on ORRs.
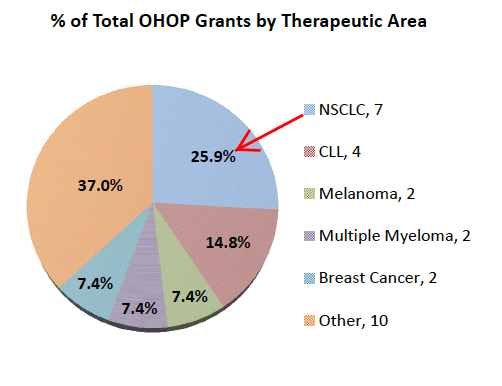
Historically, the FDA highlights that ORRs for SPA/BTD grants in oncology indications without available therapies had an Average ORR = 54% and Median ORR = 52%. For Drugs receiving BTDs in NSCLC without available therapies (e.g. T790m+) ORRs ranged from 44% to 69% with one single outlier of 29%. This compares to Tagrisso's 59% confirmed ORR vs. Roci's 28-34%. Importantly, NSCLC was the largest single tumor type receiving any special protocol assessments.
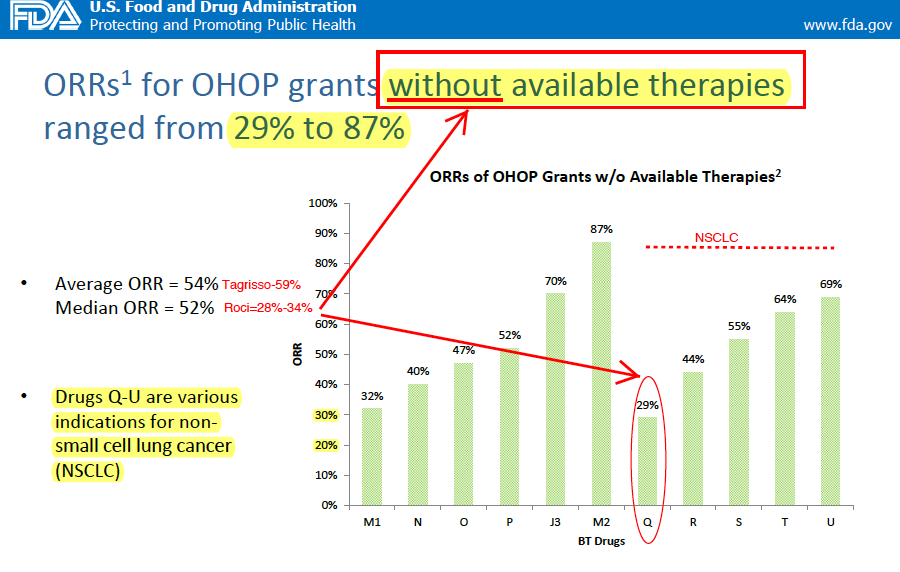
We believe the regulatory hurdle for approval since Tagrisso's approval dictates that the FDA can make a strong case that Rociletinib's current data and NDA are insufficient for approval inciting a CRL on the following grounds:
- Incomplete data

- Efficacy data too preliminary
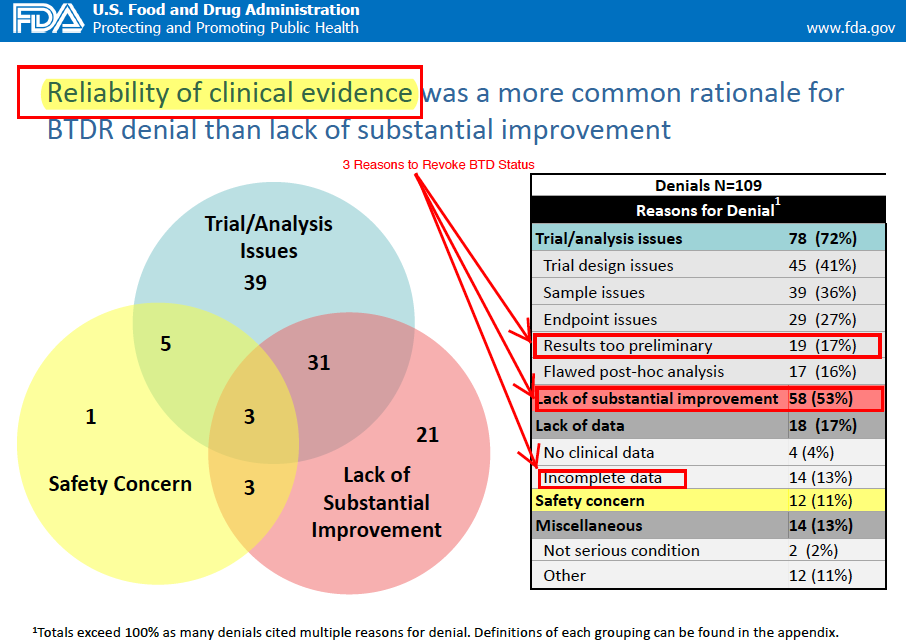
- Trial analysis issues

- Lack of substantial improvement
- Average and median ORR over best available therapy
-

- Q&A with CLVS on November 18, 2015:
- Partnered with Roche, does this adverse development alter anything there?
- No changes, planned enrollment starting 4Q15/1Q16
- When is the first CLVS and Roche/MRK data? When did they receive this notice from the FDA?
- Most likely first combo data available at ESMO or SNO in September and November respectively.
- When did CLVS get the confirmed response rate data in-house?
- End of October, 90-days after NDA.
- What data did CLVS send to the FDA exactly in the Major Amendment to NDA package?
- FDA requested "line-listings" of patient-by-patient data.
- Is Phase 3 needed for approval now, and the 625mg BID dose is now the go forward dose correct? What kind of treatment durations should we expect with Roci in 2nd and 3rd line setting now…
- FDA will determine optimal dosing on label. Treatment durations of 9-months were in the 2nd/3rd line Tiger-X population (all T790m+).
- Do you still expect front line approval in the future?
- All programs are continuing as planned.
- This in our view could be unnecessary cash burn.
- What the hypothesis here on the disparity >20% between unconfirmed and confirmed ORRs? What were your expectations?
- No hypotheses or consensus on why or how the disparity was as wide as it was. Many patients had very advanced disease.
- Unconfirmed responses of 32-34%, that upon reevaluation for confirmed responses were 27-29%, below the 30% threshold for RECIST criteria for confirmed response.
- Rociletinib, in the patients that failed to have a confirmed response, still provided "good below the neck disease control."
- Pat said that there were 40-50% of patients in the Tiger-X trial had CNS mett's prior to initiating Rociletinib correct?
- Yes. Very advanced disease.
- Do you guys need to "right-size" your commercial team for the current market opportunity?
- They have not been told by the FDA that this drug is not "approvable" and will not cut employees until CRL, or new PDUFA date is set, if a new one is set.
Disclaimer & Terms of Use
Please read out terms and conditions carefully. The use, review and accessing of the information and research contained herein and/or subsequently provided to you in a report or letter via email (the "Content") from Alpha BioPharma Advisers LLC ("Alpha BioPharma") constitutes your acceptance of these Terms & Conditions.
1. Acceptance. By using Alpha BioPharma, you are indicating your acceptance of our Terms & Conditions, and your use of the Content is subject to your compliance with these Terms & Conditions. Your right to use the Content is also subject to the terms and conditions of any additional written agreement you may have with Alpha BioPharma.
2. No Investment Advice. Alpha BioPharma is not a registered securities broker-dealer nor a registered investment adviser. Use of the Content is for your informational and educational purposes only. By viewing the Content you understand and agree that nothing contained therein constitutes a recommendation that any particular security, portfolio of securities, transaction or investment strategy is suitable for any specific person. Neither Alpha BioPharma nor any of our staff recommends that you personally buy, sell, or hold any security or portfolio of securities, or consummate any transaction or investment strategy. We do not offer investment advice, personalized or otherwise. We do not trade any securities on your behalf, nor do we offer, sell or solicit the offer or sale of any securities. The use of the Content is at your own risk. You understand that an investment in any security is subject to a number of risks, and that discussions of any security published in the Content will not contain a list or description of relevant risk factors. Alpha BioPharma recommends that you conduct your own research and due diligence and consult a qualified financial professional of your choosing for personalized advice about your financial situation before making any investment decision with respect to securities or other investment topics covered in the Content.
3. Copyright and Limitations on Use. You acknowledge that all proprietary rights in the Content are and shall remain the sole and exclusive property of Alpha BioPharma and/or its affiliates, licensors and independent third party information and content providers, and that you have no right or interest in the Content other than the right to access and to use the Content in accordance with these Terms & Conditions. You may not use the Content for any commercial purpose or repackage or redistribute the Content in any way. You agree not to infringe upon or violate the copyrights or other proprietary rights of Alpha BioPharma and/or its affiliates, licensors and independent third party information and content providers.
You may not use any automated computer program to access or retrieve the Content from the Company's website or otherwise. In addition, you may not copy, redistribute, disclose, forward (by e-mail or otherwise), furnish or sell any of the Content to any third party. All rights not granted hereunder are expressly reserved to Alpha BioPharma and/or its affiliates, licensors and independent third party information and content providers.
4. Third Party Websites, Services and Software. The Content may link to, or promote, web sites or services from other companies. You agree that Alpha BioPharma is not responsible for, and does not control, those websites and services.
5. Compliance with Applicable Law. You will comply with all applicable laws and regulations relating to the use of the Content.
6. Limitation of Liability; Disclaimers of Warranty. Alpha BioPharma is not a registered securities broker-dealer nor a registered investment adviser. You understand that the Content is furnished for your informational and educational purposes only. You may not redistribute or repackage any of the Content or market any product containing the Content for any commercial purpose. You understand that no mention of a particular security in the Content constitutes a recommendation to buy, sell, or hold that or any other security, or that any particular security, portfolio of securities, transaction or investment strategy is suitable for any specific person. You further understand that Alpha BioPharma and the Content we provide do not and will not advise you personally concerning the nature, potential, value or suitability of any particular security, portfolio of securities, transaction, investment strategy or other matter. Information regarding trading and investment as provided by Alpha BioPharma is impersonal and not tailored to the investment needs of any specific person. You acknowledge that you are responsible for your own financial and investment decisions. You should assume that as of the publication date of any of the Content Alpha BioPharma, as well as its affiliates, members, managers, officers, employees, agents, consultants, partners and licensors (collectively, the "Alpha BioPharma Parties"), has a position in all securities (and/or options of such securities) covered therein that is consistent with the position set forth in the Content. Following the publication of any of the Content, Alpha BioPharma may continue transacting in the securities covered therein, and we may be long, short, or neutral thereafter regardless of our initial recommendation. Alpha BioPharma does not intend to do business with companies covered in the Content unless otherwise noted. However, Alpha BioPharma may hold an investment position long or short with the companies mentioned in the Content, and may change its positioning without prior notice. As a result, investors should be aware that Alpha BioPharma may have a conflict of interest that could affect the objectivity of its reports.
7. Accuracy of Information. To the best of our knowledge and belief, all information contained in the Content, including but not limited to performance data and other calculations using such data, is accurate and reliable, and all information has been obtained from public sources we believe to be accurate and reliable, and not from company insiders or persons who have a relationship with company insiders. However, neither Alpha BioPharma nor any of the Alpha BioPharma Parties warrant or guarantee that: (i) the Content is accurate, reliable or correct; (ii) that the Content will be available at any particular time or location; or (iii) that any defects or errors in the Content will be corrected. As markets change continuously, previously published information and data may not be current and should not be relied upon. All information in the Content is presented only as of the date published or indicated, and may be superseded by subsequent market events or for other reasons. The Content Is Provided "As Is" And "As Available" And Alpha Biopharma Makes No Warranty Of Any Kind, Express, Implied Or Statutory, Including, But Not Limited To, Any Warranties Of Accuracy, Completeness, Timeliness, Merchantability, Fitness For A Particular Purpose, Or Noninfringement .
8. Disclaimer. Use of the content is solely at your risk. Alpha biopharma and the alpha biopharma parties will not be liable to you or any other person as a result of your use of, or reliance on, or inability to use the content for indirect, consequential, special, incidental, punitive, or exemplary damages, including, without limitation, lost profits, lost savings and lost revenues, whether or not characterized in negligence, tort, contract, or other theory of liability, even if alpha biopharma or any of the alpha biopharma parties have been advised of the possibility of or could have foreseen any of the excluded damages, and irrespective of any failure of an essential purpose of a limited remedy. If any applicable authority holds any portion of this section to be unenforceable, then alpha biopharma or the alpha biopharma parties' liability will be limited to the fullest possible extent permitted by applicable law.
Nov. 27, 2015 Alpha BioPharma Advisers LLC
Supporting Documents